Lesch–Nyhan Syndrome: Mrna Expression Of Hprt In
Di: Ava
Eight Lesch-Nyhan disease and variant cases recapitulate the effects of HPRT deficiency on cell-fate marker expression and OXPHOS To externally validate the HPRT1 KO data in people affected with disease and to demonstrate molecular effects on different genetic backgrounds, we made DA NPCs from three LND subjects, five LNV subjects Nguyen KV, Naviaux RK, Paik KK, et al. (2012) Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of DNA.
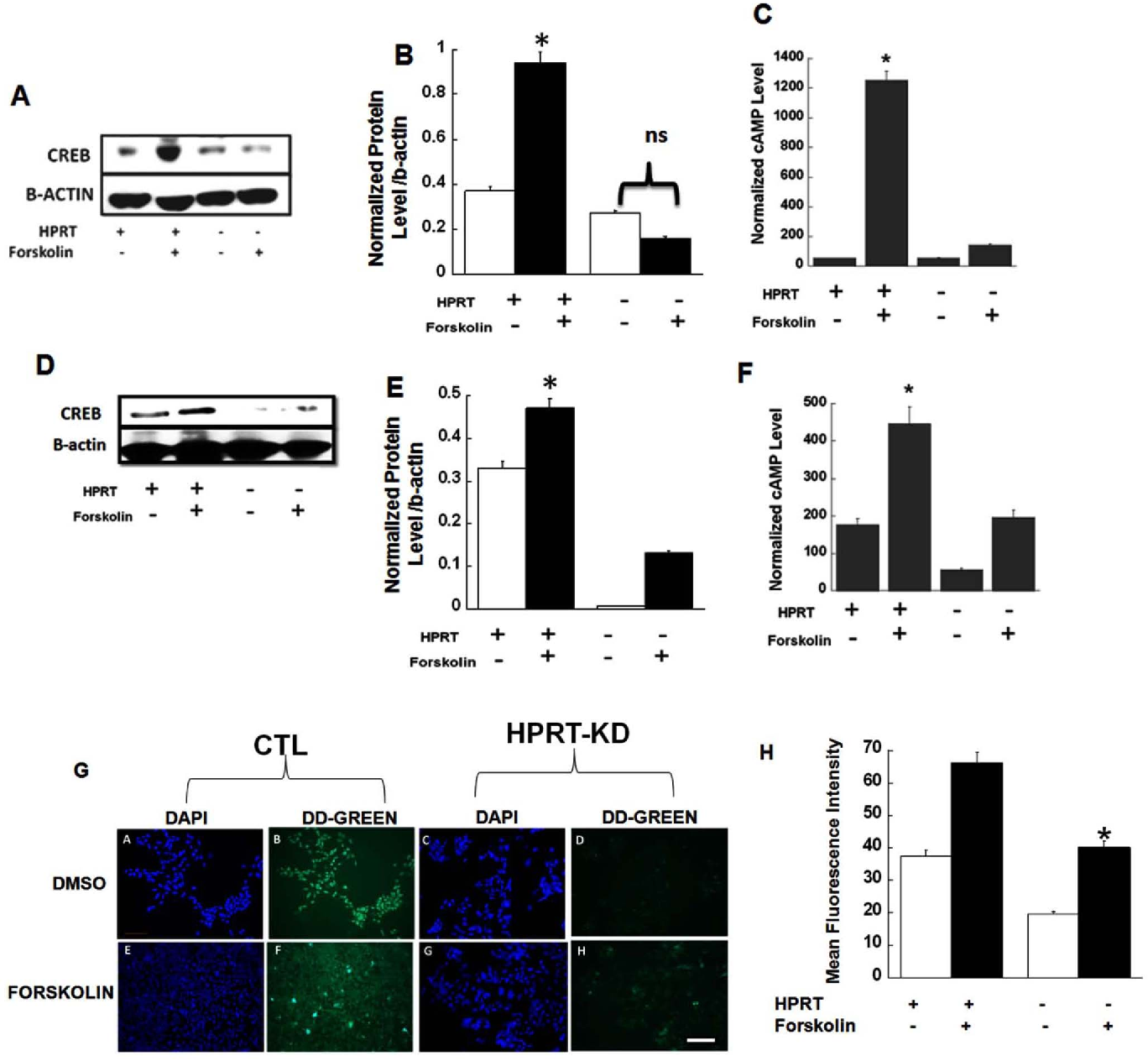
With this is mind, HPRT has the potential to become a significant biomarker not only for the characterization of cancer, but also for its potential treatment. Keywords: Hypoxanthine guanine phosphoribosyltransferase (HPRT or HGPRT); Kelley–Seegmiller syndrome; Lesch–Nyhan syndrome; Salvage enzymes.
Das Lesch-Nyhan-Syndrom (LNS), auch unter den Synonymen Hyperurikämie -Syndrom und Hyperurikose bekannt, ist eine Stoffwechselerkrankung als Folge eines Gendefektes, der X-chromosomal-rezessiv vererbt wird. Das Lesch-Nyhan-Syndrom, kurz LNS, ist eine seltene X-chromosomal-rezessiv vererbte Stoffwechselerkrankung aus dem rheumatischen Formenkreis.
Normal HPRT coding region in complete and partial HPRT deficiency
19 Nguyen KV, Naviaux RK, Paik KK, Nyhan WL: Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of the DNA. Lesch–Nyhan Syndrome: mRNA expression of HPRT in patients with enzyme proven defi ciency of HPRT and normal HPRT coding region of the DNA. Mol Gen Metab. 2012;106(4):498–501. https://doi. org In the present study we present four patients with partial HPRT deficiency and one patient with Lesch–Nyhan syndrome who showed a normal HPRT coding sequence and markedly decreased HPRT mRNA expression. This is the first report of a patient with Lesch–Nyhan syndrome due to a defect in HPRT gene expression regulation.
In 1964, Lesch and Nyhan provided the first detailed clinical description of Lesch-Nyhan syndrome with a report of two brothers with hyperuricemia and a characteristic neurobehavioral syndrome that included motor dysfunction and self-injurious behavior. The To elucidate the molecular basis of neurological disorders in the Lesch-Nyhan syndrome and to devise a method for delivering genetic information into the appropriate cells of brain tissue at the appropriate time, it is essential to understand the molecular mechanism responsible for the human HPRT gene expression, especially in the
The human HPRT1 gene is located on the X-chromosome (Xq26.3). This gene has only one functional mRNA transcript that encodes the enzyme hypoxanthine-guanine phosphoribosyltransferase. 1 It is one of the key enzymes in the salvage pathway of purine synthesis. Mutation in the HPRT1 gene is the most common reason underlying Lesch–Nyhan
- A review of HPRT and its emerging role in cancer
- The Study on the Clinical Phenotype and Function of HPRT1 Gene
- Induced pluripotent stem cells from subjects with Lesch-Nyhan disease
- Clinical utility gene card for: Lesch–Nyhan syndrome
Hypoxanthine-guanine phosphoribosyltransferase (HPRT, EC 2.4.2.8) is a purine salvage enzyme that catalyses the conversion of hypoxanthine and guanine to their respective mononucleotides. Partial deficiency of this enzyme can result in the overproduction of uric acid leading to a severe form of gout, whilst a virtual absence of HPRT activity causes the Lesch-Nyhan syndrome
La enfermedad de Lesch-Nyhan es una enfermedad genética considerada dentro de las enfermedades raras, caracterizada por un déficit en la función de la enzima hipoxantina-guanina fosforribosil transferasa (HPRT). Los pacientes afectados presentan hiperuricemia, trastornos motores, retraso mental y, en los casos más graves, automutilaciones. Las Lesch–Nyhan disease (LND) is associated with a complete deficiency of hypoxanthine-guanine phosphoribosyltransferase (HPRT) activity due to mutations in the HPRT1 gene. Although the physiopathology of LND-related neurological manifestations remains unknown, a defective neuronal developmental process is the most widely accepted hypothesis. In the present study we present four patients with partial HPRT deficiency and one patient with Lesch-Nyhan syndrome who showed a normal HPRT coding sequence and markedly decreased HPRT mRNA
Lesch-Nyhan variant syndrome: real-time RT-PCR for mRNA quantification in variable presentation in three affected family members Nucleosides Nucleotides Nucleic Acids, 31 (2012), pp. 616 – 629 A number sign (#) is used with this entry because Lesch-Nyhan syndrome (LNS) is caused by mutation in the HPRT gene (308000), which encodes hypoxanthine guanine phosphoribosyltransferase, on chromosome Xq26. Most HPRT-deficient patients, biochemically diag-nosed by a null HPRT activity in erythrocytes, present HPRT mRNA expression, and molecular diagnosis can be accomplished by RNA extraction, reverse
Complete deficiency of the purine salvage enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT) results in a devastating neurological disease, the Lesch-Nyhan syndrome. This disorder has been identified as a candidate for initial attempts at somatic cell gene therapy. We have previously reported the construction of a recombinant herpes simplex virus Lesch-Nyhan Disease (LND) is a rare X-linked recessive metabolic and neurological syndrome due to the deficiency of hypoxanthine-guanine phosphoribosyltransferase (HPRT). Besides its well known “housekeeping” function this purine salvage enzyme has revealed an unexpected role in neurodevelopment, unveiled by the peculiar neurological symptoms flanking hyperuricemia in

Abstract Purpose: Lesch-Nyhan syndrome (LNS) is an X-linked recessive disorder of purine metabolism with a defect in the activity of hypoxanthine-guanine phosphoribosyl transferase (HPRT). The aim of this study was to describe 2 cases of LNS, who were very dificult to be diagnosed. Lesch-Nyhan disease (LND) is an X-linked genetic disorder caused by mutations of the hypoxanthine guanine phosphoribosyltransferase (HPRT) purine biosynthesis gene and characterized by aberrant
RESUMO Síndrome de Lesch-Nyhan é uma desordem genética rara caracterizada pela deficiência da atividade enzimática da hipoxantina guanina fosforribosiltransferase (HPRT), caracterizada por hiperuricemia, hiperuricosúria e diversas alterações neurológicas concomitantes, principalmente distúrbios do movimento, déficit cognitivo e auto-mutilação. Os
title Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of the DNA.(English) 1 reference stated in 22766437 8 December 2017 main subject Lesch-Nyhan syndrome 0 references author Robert K Naviaux series ordinal 2 object named as Robert K Naviaux 1 reference 22766437 8
Because the HPRT gene is on the X-chromosome, males are affected and females in the families are atrisk of being carriers of the mutation. We report two LNS affectedmembers of a family in whom a normal HPRT coding sequence wasassociated with markedly decreased mRNA expression of HPRT.2. Materials and methods2.1.
La enfermedad de Lesch-Nyhan es una enfermedad genética considerada dentro de las enfermedades raras, caracterizada por un déficit en la función de la enzima hipoxantina-guanina fosforribosil transferasa (HPRT). Los pacientes afectados presentan hiperuricemia, trastornos motores, retraso mental y, en los casos más graves, automutilaciones. Las Lesch-Nyhan disease is a rare genetic disease characterized by a deficiency in the function of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). Patients affected by this disease experience hyperuricemia, motor disorders, mental retardation and, in the most severe cases, self-mutilation. Its clinical manifestations depend on the enzymatic activity
Inherited mutation of the purine salvage enzyme, hypoxanthine guanine phosphoribosyltransferase (HPRT) gives rise to Lesch-Nyhan syndrome (LNS) or Lesch-Nyhan variants (LNV). We report a case of two LNS affected members of a family with deficiency of activity of HPRT in intact cultured fibroblasts in whom mutation could not be found in the HPRT
Lesch-Nyhan syndrome (LNS) is a neurodevelopmental disorder caused by mutations in the gene encoding the purine metabolic enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT). A series of motor, cognitive and neurobehavioral anomalies Gibbs and Caskey (1987) used the ribonuclease A cleavage procedure, with a polyuridylic acid-paper affinity chromatography step, to identify the mutation lesions in the HPRT mRNA of patients with Lesch-Nyhan syndrome (LNS; 300322).
In Lesch–Nyhan syndrome and Kelley–Seegmiller syndrome, the regulatory nature of HPRT is demonstrated as the lack of the protein results in an overproduction of purines. Purpose: Lesch-Nyhan syndrome (LNS) is an X-linked recessive disorder of purine metabolism with a defect in the activity of hypoxanthine-guanine phosphoribosyl transferase (HPRT). The aim or his study was to describe 2 cases of LNS, who were very difficult to be diagnosed. Methods: Two cases of LNS were described: 6-month-old and 13-month-old
Lesch-Nyhan syndrome (LNS) is an X-linked recessive disorder caused by mutations in the HPRT1 gene. The clinical features and mutation spectrum of 26 Korean LNS patients from 23 unrelated families were retrospectively reviewed. The HPRT1 gene was analyzed by direct sequencing of genomic DNA. The median age at diagnosis was 2.3 years (range, 4
- Lesen Sie 209 Bewertungen Zu Auronia.De
- Letzte Generation Von Kostas Koufogiorgos
- Let’S Play Gothic 2 Dndr _ Gothic 2 Let’S Play
- Les Donnees Structurees _ Chapitre 2 les données structurées et leur traiteme
- Les Parcs De Jeux Couverts Pour Enfants À Paris Et En Ile De France
- Lette-Akademie Berlin Gemeinnützige Gmbh
- Les Marseillais Vs Le Reste Du Monde 6: Le Casting Complet Dévoilé
- Letesha Marrow Age, Biography, Facts, Net Worth, Career
- Les Meilleurs Restaurants Avec Menu De Noël 2024 À Arcachon
- Les Différences Entre Le Solaire Thermique Et Photovoltaïque